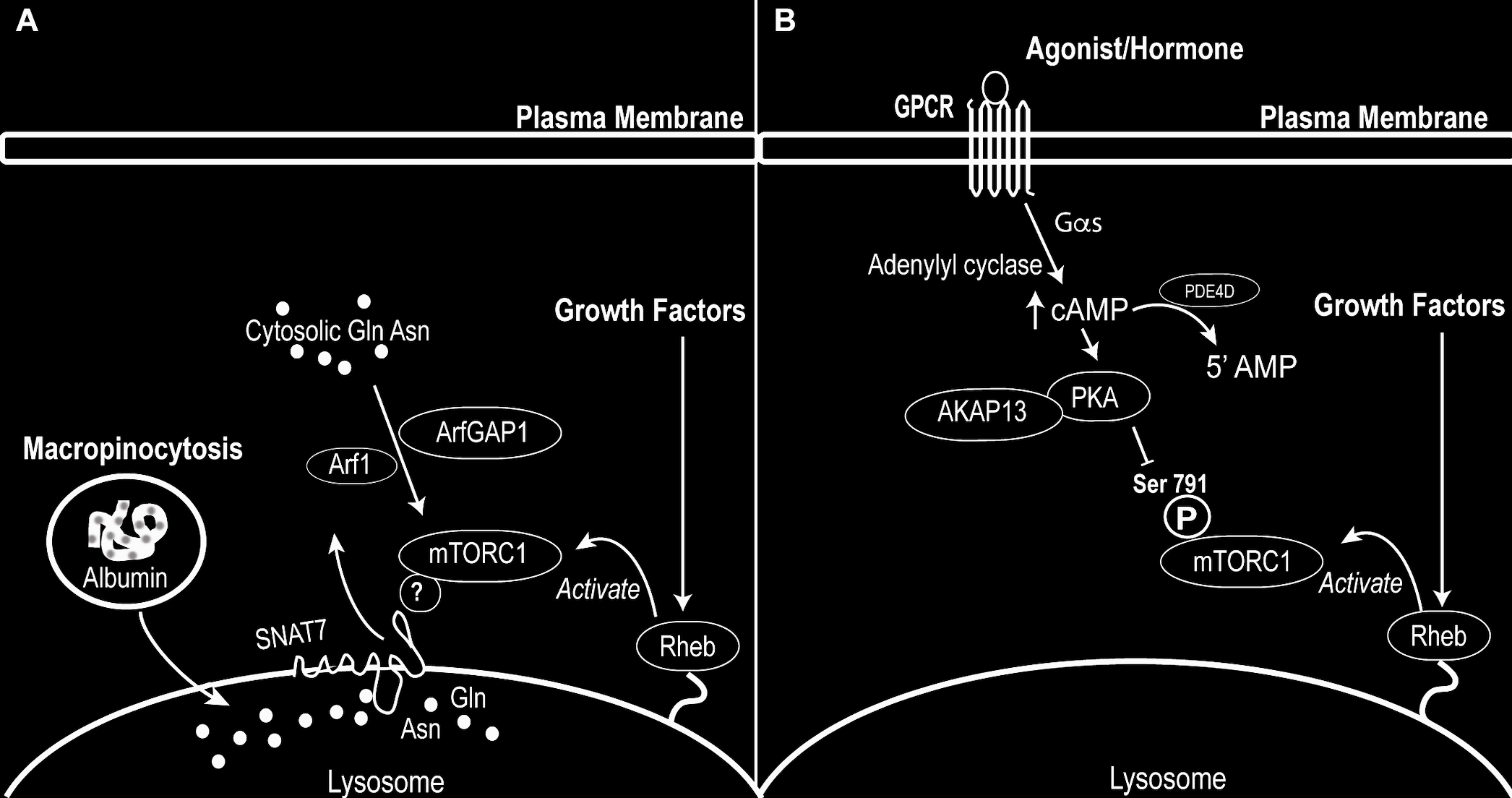
Nutrients are sensed by cells to regulate growth, metabolism, and autophagy, and disruption of these processes contributes to human disease. The mammalian target of rapamycin (mTOR) is a conserved serine/threonine kinase and the catalytic component of mTOR complex 1 (mTORC1), a central signaling hub that integrates nutrient availability to coordinate these cellular responses. Often described as a master regulator of cell growth, mTORC1 promotes anabolic processes such as protein synthesis while suppressing catabolic pathways, including autophagy.
The Jewell laboratory is interested in understanding how mTORC1 activity is controlled by upstream signals. Our research focuses on two major areas: (1) activation of mTORC1 in response to amino acids, particularly glutamine (Gln) and asparagine (Asn) (Figure A), and (2) regulation of mTORC1 by G protein–coupled receptor (GPCR) signaling (Figure B). In particular, we investigate GPCRs coupled to Gαs proteins and their role in inhibiting mTORC1 signaling.
mTORC1 activity is elevated in a wide range of human diseases, including cancer, yet current therapies targeting this pathway have shown limited clinical benefit. By defining the molecular mechanisms that regulate mTORC1, our work aims to identify new strategies for more effective therapeutic intervention.